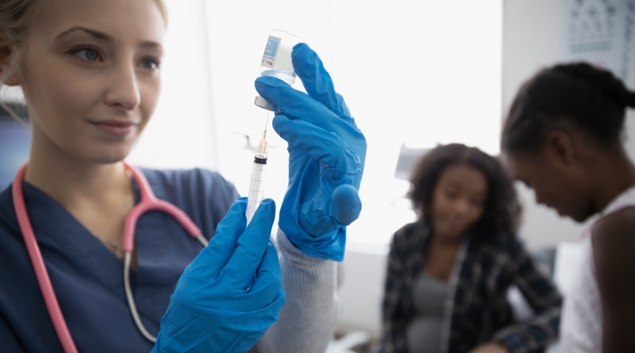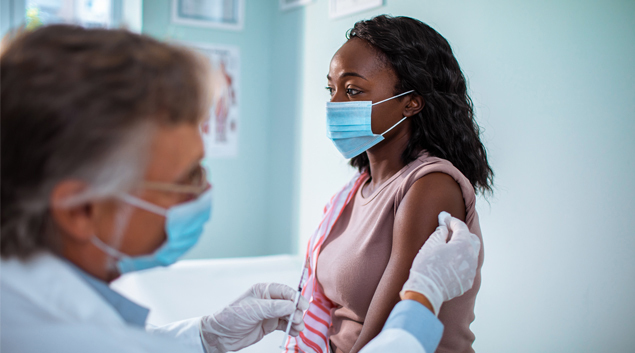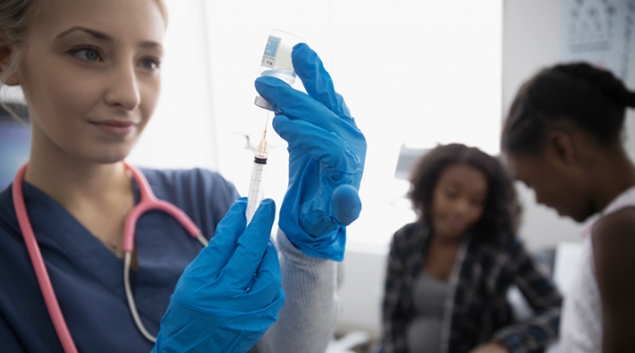
On Monday, the U.S. Food and Drug Administration granted Emergency Use Authorization for Eli Lilly and Company’s investigational neutralizing antibody bamlanivimab. Bamlanivimab is authorized for the treatment of mild to moderate COVID-19 in adults and pediatric patients 12 years and older with a positive COVID-19 test who are at high risk for progressing to severe COVID-19 and/or hospitalization.
Bamlanivimab should be administered as soon as possible after a positive COVID-19 test and within 10 days of symptom onset. The authorization allows for the distribution and emergency use of the antibody treatment, which is administered via a single intravenous infusion.
David A. Ricks, Lilly’s chairman and CEO, said in a statement that the EUA allows the company to make the treatment available for recently diagnosed, high-risk patients and will be a “valuable tool” for doctors fighting the increasing burden of the pandemic.
HIMSS20 Digital
Learn on-demand, earn credit, find products and solutions. Get Started >>
WHAT’S THE IMPACT?
The EUA is based on data from BLAZE-1, a randomized, double-blind, placebo-controlled Phase 2 study in patients with recently diagnosed mild to moderate COVID-19 in the outpatient setting. Patients treated with bamlanivimab showed reduced viral load and rates of symptoms and hospitalization.
In BLAZE-1, frequency and types of adverse events were similar between bamlanivimab and placebo, with the majority being mild to moderate in severity. Infusion reactions and other allergic hypersensitivity events have been reported. The EUA includes a warning for hypersensitivity including anaphylaxis and infusion-related reactions.
Dr. Daniel Skovronsky, Lilly’s chief scientific officer and president of Lilly Research Laboratories, said data shows that bamlanivimab can help patients clear the virus when given early in the course of the disease, which may reduce COVID-19-related hospitalizations. That, said Skovronsky, supports the belief “that neutralizing antibodies can be an important therapeutic option for patients fighting this virus.”
The FDA grants emergency use authorization to provide availability of a medicine that may help diagnose, treat or prevent a life-threatening disease when no adequate and approved alternatives are available. This use of bamlanivimab is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use, unless the authorization is terminated or revoked sooner.
The authorization is temporary, and does not replace the formal review and approval process. Bamlanivimab remains an investigational drug and has not been approved under a Biologics License Application. Evaluation of its safety and efficacy is ongoing across a range of patient populations impacted by COVID-19. Data from these studies will be used to support a future BLA submission involving bamlanivimab.
Lilly will begin shipping bamlanivimab immediately to AmerisourceBergen, a national distributor, which will distribute it as directed by the U.S. government’s allocation program.
ALLOCATION AND MANUFACTURING
The U.S. government has purchased 300,000 doses of bamlanivimab and committed that Americans will have no out-of-pocket costs for the medicine, although healthcare facilities may charge a fee for the product’s administration.
The federal government is responsible for the appropriate allocation of the treatment. Weekly allocation decisions will be proportionally based on confirmed COVID-19 cases in each state and territory over the previous seven days, based on data from the U.S. Department of Health and Human Services’ Protect data collection platform.
Each week, state and territorial health departments will select sites of care – that are accessible and can minimize infection transmission – to receive allocated doses and will provide AmerisourceBergen the list of sites. Sites of care will then confirm their need, and AmerisourceBergen will distribute bamlanivimab overnight.
To ensure rapid access of the treatment, Lilly has invested in large-scale manufacturing of bamlanivimab at risk – even before data demonstrated its potential to become a meaningful therapeutic option for COVID-19. Lilly has a global supply chain in place to produce bamlanivimab, with numerous manufacturing sites worldwide.
Lilly anticipates manufacturing up to one million doses of 700 mg by the end of 2020, for use around the world through early next year. Beginning in Q1 2021, the supply of Lilly’s antibody therapy is expected to increase substantially, as additional manufacturing resources come online throughout the year.
Lilly is in discussions with global regulators to make bamlanivimab available around the world. Global allocation will be made based on Lilly’s guiding principles that aim to ensure access for patients with high unmet need.
THE LARGER TREND
In October, the FDA approved the antiviral drug Veklury (remdesivir) for use in adult and pediatric patients 12 years old and older and weighing at least 88 pounds for COVID-19 treatments requiring hospitalization.
Veklury should only be administered in a hospital or in a healthcare setting capable of providing acute care comparable to inpatient hospital care, the FDA said. Veklury is the first coronavirus treatment to receive FDA approval.
In order to ensure continued access to the pediatric population previously covered under the EUA, the FDA revised it to authorize Veklury’s use for treatment of suspected or laboratory-confirmed COVID-19 in hospitalized pediatric patients weighing 7.7 lbs. to less than 88 lbs., or hospitalized pediatric patients less than 12 years old weighing at least 7.7 lbs. Clinical trials assessing the safety and efficacy of Veklury in this pediatric patient population are ongoing.
When President Trump was diagnosed with COVID-19, he was given remdesivir, as well as experimental antibody cocktail being developed by the drug maker Regeneron, in addition to several other drugs, including zinc, vitamin D and the generic version of the heartburn treatment Pepcid, according to The New York Times which cited a letter from his doctor that was released by the White House.
Under the Federal Food, Drug, and Cosmetic Act, approval of a new drug product requires substantial evidence of effectiveness and a demonstration of safety for the drug’s intended uses. In considering approval of a drug, the FDA conducts a scientific benefit-risk assessment to ensure that the product’s benefits outweigh its risks for the intended population. This is different from the standard used in the issuance of an EUA.
ON THE RECORD
“Authorization of this new Eli Lilly antibody treatment is a significant step forward in treating patients and bridging us to the rollout of safe and effective vaccines, with all of these efforts made possible by Operation Warp Speed,” said HHS Secretary Alex Azar. “Operation Warp Speed is helping to ensure that therapeutics like Lilly’s can reach patients without a day wasted.”